SInCre
An Open Source Integrated Computational Resource for the Analysis of the Structural Interactome to predict Off-Site Interactions of Drug Candidates
Background
Knowledge of molecular interactions that define various cellular processes and form crucial inter-connections among them will be central to our understanding of cellular function and will bear a direct influence on drug discovery. In the current drug discovery pipelines, there is uncertainty in many steps due to glaring knowledge gaps and lack of appropriate data as well as methods to bridge such gaps. Many new medicines fail because they exhibit off-target binding. This can lead to harmful side effects, which are often not discovered until late in the research development programme, when millions of dollars may have been invested in the new drug. A major and very urgent challenge for experimental and computational biologists and chemists is to discover ways of identifying off-target sites for drugs at an early stage in the research programme. Detailed structural knowledge of the interactions between molecules in the cell may provide one way of approaching this problem. The objective would be to define the structural interactome, an inventory of the various interactions between macromolecules and both natural and synthetic small molecules. To achieve this objective, integrated databases are required to bring information on protein sequences and structures of families and superfamilies together with that on interactions with other proteins, nucleic acids, polysaccharides, and small molecules. Efficient tools are required to search these databases in order to identify binding sites for small-molecule drug candidates that might lead to off-target binding and unintended harmful effects. For maximum impact on the development of new medicines, particularly tuberculosis, freely available, open-source data and tools are required by researchers throughout the world. The integrated suite of databases available through this resource provides a structural dimension to studies on tuberculosis from several perspectives ranging from structure based drug target and druggability assessments on one hand to the analysis of genetic variation in relation to disease and drug discovery on the other. Identification of functional domains using sensitive approaches, remote homology detection, genome-scale structural modelling or the structural proteome by employing multiple algorithms, identification of small-molecule binding pockets and the characterization of the binding pocketome, structure-based function annotation, identification of small-molecule binders, protein-protein interactions and possible shortlist of binders from the FDA approved drug databases, natural compounds that bind to important proteins in the pathogen and to protein-protein interfaces, are all features of this resource and form necessary prerequisites for identification of off-target binding.
Individual resources, tools and algorithms used for generating this data are described briefly below.
Profile-based domain assignment
Three sensitive profile based techniques were used to achieve enhanced domain annotation for the proteins encoded in the pathogen using structural (SCOP) and sequence (Pfam) databases. Each protein sequence was subjected to all the three approaches, as shown in the figure, to achieve a reasonably good sequence coverage for function annotation. Each of the domain family associations made were evaluated in terms of their statistical significance, as described in the parameters section of the figure. For domain assignments using Pfam database, gathering threshold cut-offs were used to extract reliable hits. For hits identified by HHblits, e-value <= 0.001 was used and RPS-BLAST based search, e-value <=0.01 was used. Another criteria i.e. profile coverage, of 70% cut-off (70% of the domain family profile aligns significantly with the protein sequence) was employed in order to refine the domain assignments made. All the domain assignments were manually curated to ensure reliability.
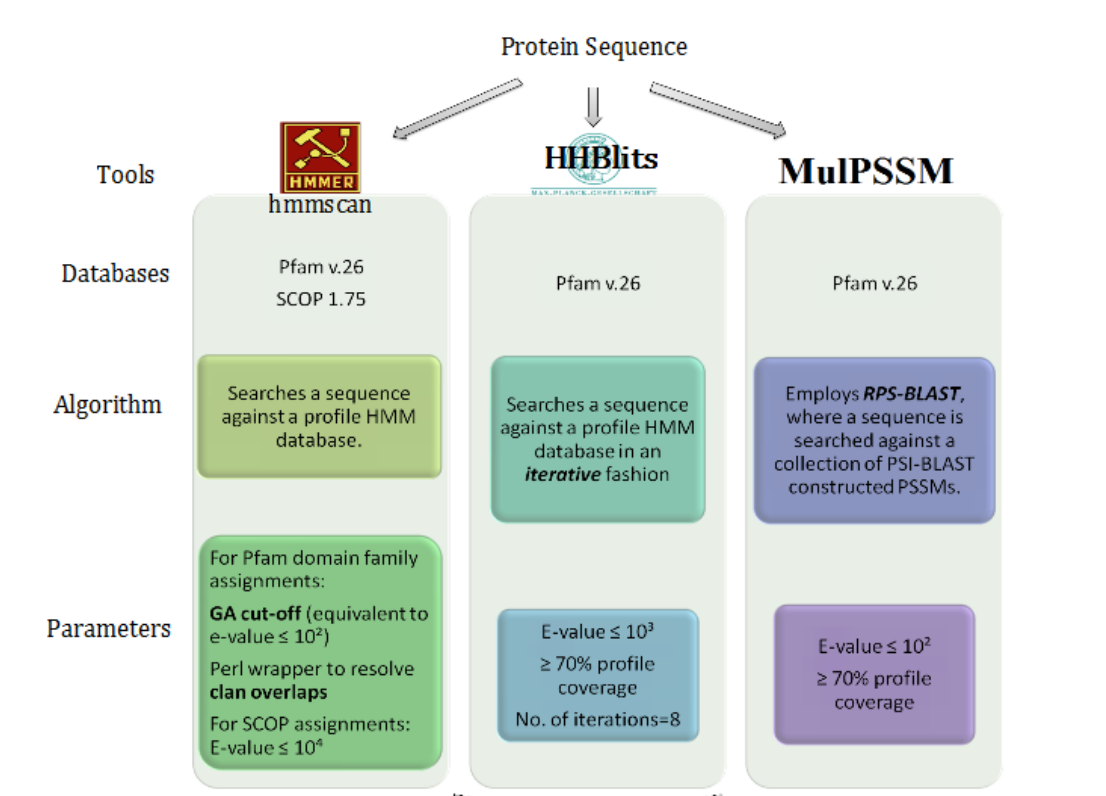
Sequences with no domain assignment were subjected to a fold recognition approach PHYRE2 to explore the secondary structure elements in the unannotated protein sequences. Further, protein sequences with no reliable information on the fold adopted, were scanned against non-redundant database using HHblits (e-value <=0.001).
References:
MulPSSM
V. S. Gowri, O. Krishnadev, C. S. Swamy, and N. Srinivasan. MulPSSM: a database of multiple position-specific scoring matrices of protein domain families
Nucl. Acids Res. (2006) 34 (suppl 1): D243-D246 doi:10.1093/nar/gkj043
AlignHUSH
O. Krishnadev and N. Srinivasan. AlignHUSH: Alignment of HMMs using structure and hydrophobicity information.
BMC Bioinformatics (2011), 12:27 doi:10.1186/1471-2105-12-275
targetTB
targetTB is a comprehensive in silico target identification pipeline, targetTB, for Mycobacterium tuberculosis. The pipeline incorporates a network analysis of the protein-protein interactome, a flux balance analysis of the reactome, experimentally derived phenotype essentiality data, sequence analyses and a structural assessment of targetability. Using flux balance analysis and network analysis, proteins critical for survival of M. tuberculosis are first identified, followed by comparative genomics with the host, finally incorporating a novel structural analysis of the binding sites to assess the feasibility of a protein as a target. Further analyses include correlation with expression data and non-similarity to gut flora proteins as well as 'anti-targets' in the host, leading to the identification of 451 high-confidence targets. Through phylogenetic profiling against 228 pathogen genomes, shortlisted targets have been further explored to identify broad-spectrum antibiotic targets, while also identifying those specific to tuberculosis. Targets that address mycobacterial persistence and drug resistance mechanisms are also analysed.
Reference:
Raman, K., Yeturu, K., Chandra, N. (2008) targetTB: a target identification pipeline for Mycobacterium tuberculosis through an interactome, reactome and genome-scale structural analysis. BMC Syst Biol, 2, 109.
PURE (Prediction of unassigned regions)
In the continuing interest to identify target genes for efficient drug discovery, we searched for interesting targets that are likely involved in host-pathogen interactions (enzymes and lipid modifying proteins) and not contain close human counterparts. The well-annotated genes would have been considered in screening of gene targets by other groups and we chose to look into unannotated genes in the genome of Mycobacterium tuberculosis(H37Rv strain). Domain finding of unannotated genes was pursued using a computationally intensive bioinformatics pipeline called PURE (Prediction of Unassigned REgions) that has been developed earlier in the group (Reddy et al., Nature Protocols (2008;http://www.ncbi.nlm.nih.gov/pubmed/18554415) and exploits domain association through homology. Direct identification and association of sequence-based domains from Pfam database using HMMSCAN program from HMMER suite left behind 566 full-length sequences with no domain associations at all. We could find 15 Pfam domain associations with the initial set of parameters in PURE and an additional 279 domains through relaxed thresholds. 7 hits, out of 15 associations obtained by strict thresholds, had to be discarded upon closer scrutiny of the connecting sequences of homologues identified since they had a substantial overlap with human proteins. Structure prediction and structural modls could be generated for eight of the 13 hits, which are interestingly enzymes. Virtual screening will be pursued for the eight “targets”, which we are excited about and strongly recommend for function-test experiments by the other groups in the OSDD consortium.
CREDO
A protein-ligand interaction database for drug discovery
Harnessing data from the growing number of protein-ligand complexes in the Protein Data Bank (PDB) is an important task in drug discovery. In order to benefit from the abundance of three-dimensional structures, structural data must be integrated with sequence as well as chemical data and the protein-small molecule interactions characterised structurally at the inter-atomic level. Here, we present CREDO, a new publicly available database of protein-ligand interactions, which represents contacts as structural interaction fingerprints, implements novel features and is completely scriptable through its application programming interface (API). Features of CREDO include implementation of molecular shape descriptors with Ultrafast Shape Recognition (USR), fragmentation of ligands in the PDB, sequence-to-structure mapping and the identification of approved drugs. Selected analyses of these key features are presented to highlight a range of potential applications of CREDO. We believe that the free availability and numerous features of CREDO database will be useful not only for commercial but also for academia-driven drug discovery programmes.
Reference
Schreyer AM & Blundell TL (2013) CREDO: a structural interactomics database for drug discovery.
Database The Journal of Biological Databases and Curation. 01/2013; 2013:bat049. DOI:10.1093/database/bat049
http://marid.bioc.cam.ac.uk/credo
TIMBAL
TIMBAL holds small molecules disrupting protein-protein interactions
To date, drug discovery has focused in the main part on a handful of targets that meet the "classical" druggable criteria: being linked to disease and having a beautiful pocket able to bind a small drug-like molecule (Hopkins and Groom, 2002). However the successes of monoclonal antibodies as therapeutic agents are changing the perspective of what makes a drug target. These biologicals disrupt multi-protein complexes, which are the key points of almost all processes in living organisms. Still, antibodies being expensive and difficult to administer are not the ideal drugs, and they target only extra cellular molecules. In recent years, growing evidence of the possibility of modulation of protein-protein interactions by small molecules is opening the door for new approaches and concepts in drug discovery (Whitty and Kumaravel, 2006). TIMBAL aims to be a resource which will give insights in the type of molecules favoured by protein interfaces and in the type of interactions these systems present. TIMBAL is a database containing small molecules that modulate protein-protein interactions. It was first created in 2008, by manually curating information extracted from relevant scientific publications. An analysis of the data was published in 2009, (Higueruelo et al, 2009). The growth of data in the past years makes hand-curated databases a phenomenally time-consuming task. The maintenance of TIMBAL is done now through automated searches on the ChEMBL database. The list of known PPI targets and its orthologs has been transtaled into UniProt codes. These codes are then used in ChEMBL for searching small molecule data related to these proteins in binding assays with confidence that the assay is directly assigned to a single protein or its homolog.
Reference:
Higueruelo A.P., Jubb H. and Blundell T.L. (2013) TIMBAL v2: update of a database holding small molecules modulating protein朴rotein interactions.
Database (2013) Vol. 2013: article ID bat039; doi:10.1093/database/bat039
http://www-cryst.bioc.cam.ac.uk/timbal
http://mordred.bioc.cam.ac.uk/timbal
TIBLE
TIBLE holds ligand-based off target prediction and small molecules data
There are two main approaches to predict of off-target activity. Structure-based relies in the similarity of
the targets binding pockets, whereas ligand-based connect targets based on the similarity of their
ligands. Both methodologies complement each other (Xie, 2011). TIBLE collects small molecule data (MIC for mycobacterium and
binding to isolated Mtb targets) from the ChEMBL database (Gaulton, 2011) and the CDD (Ekins,
2010). There are 75 Mtb targets with small molecule binding data. For each of these targets, three
independent algorithms SEA (Keiser, 2007;Keiser, 2009), PharmMapper (Liu, 2010) and PASS
(Poroikov, 2007) are used to derive off-target ligand-based predictions.
http://mordred.bioc.cam.ac.uk/tible
CHOPIN
http://structure.bioc.cam.ac.uk/chopin
Mtb Structural Proteome
Structural annotation of Mycobacterium tuberculosis proteome was carried out by one of the group. PDB holds total of 324 crystal structures of Mtb proteins and comparative models were generated for 2737 proteins, thus totalling up the structure availability for 70% of Mtb proteome. Structural models were generated using Modpipe, a software suite along with ModBase, a comparative protein structure model database.
Reference http://proline.physics.iisc.ernet.in/Tbstructuralannotation/Anand, P., Sankaran, S., Mukherjee, S., et al. (2011) Structural annotation of Mycobacterium tuberculosis proteome. PLoS One, 6, e27044.
Binding site detection
Binding sites for all sturctural models were detected. Computational methods for binding site detection is described below.

Drug binding site database and comparison
DrugBank and DrugPort were used to prepare a combined list of drugs or drug-like compounds, these included approved and experimental drugs and nutraceuticals. XML data files were obtained from these two databases and later parsed to extract information on proteins complexed with any of these drugs present in PDB. The binding sites were then extracted from these complexes. Residues of all atoms that lie within 4.5Å of any atom in the drug molecule were extracted as part of binding site.
10658(from Drugbank)+2516(from Drugport) drug-binding sites were obtained from PDB through this process (list is available at http://proline.biochem.iisc.ernet.in/mtbpocketome/methods.php). High-confidence targets from Mtb were scanned using these known drug-binding sites and also drug-binding sites were scanned for similarities against different binding site clusters.
Anand,P., and Chandra,N. (2014) Characterizing the pocketome of Mycobacterium tuberculosis and application in rationalizing polypharmacological target selection. Scientific Reports 2014 (In Press).



